BioAcyl Corp |
|
Chaudhari, N., Talwar, P., Parimisetty, A., & others. (2014). A Molecular Web: Endoplasmic Reticulum Stress, Inflammation, and Oxidative Stress. Front. Cell. Neurosci. 8. Added by: Dr. Enrique Feoli (18/02/2021, 18:48) Last edited by: Dr. Enrique Feoli (27/02/2021, 18:26) |
| Resource type: Journal Article Published DOI: 10.3389/fncel.2014.00213 ID no. (ISBN etc.): 1662-5102 BibTeX citation key: Chaudhari2014 View all bibliographic details |
Categories: BioAcyl Corp Subcategories: Immunometabolism Creators: Chaudhari, others, Parimisetty, Talwar Collection: Front. Cell. Neurosci. |
Views: 6/551
|
| Abstract |
|
Execution of fundamental cellular functions demands regulated protein folding homeostasis. Endoplasmic reticulum (ER) is an active organelle existing to implement this function by folding and modifying secretory and membrane proteins. Loss of protein folding homeostasis is central to various diseases and budding evidences suggest ER stress as being a major contributor in the development or pathology of a diseased state besides other cellular stresses. The trigger for diseases may be diverse but, inflammation and/or ER stress may be basic mechanisms increasing the severity or complicating the condition of the disease. Chronic ER stress and activation of the unfolded protein response (UPR) through endogenous or exogenous insults may result in impaired calcium and redox homeostasis, oxidative stress via protein overload thereby also influencing vital mitochondrial functions. Calcium released from the ER augments the production of mitochondrial Reactive Oxygen Species (ROS). Toxic accumulation of ROS within ER and mitochondria disturb fundamental organelle functions. Sustained ER stress is known to potentially elicit inflammatory responses via UPR pathways. Additionally, ROS generated through inflammation or mitochondrial dysfunction could accelerate ER malfunction. Dysfunctional UPR pathways has been associated with a wide range of diseases including several neurodegenerative diseases, stroke, metabolic disorders, cancer, inflammatory disease, diabetes mellitus, cardiovascular disease and others. In this review we have discussed the UPR signaling pathways, and networking between ER stress induced inflammatory pathways, oxidative stress and mitochondrial signaling events which further induce or exacerbate ER stress.
|
| Notes |
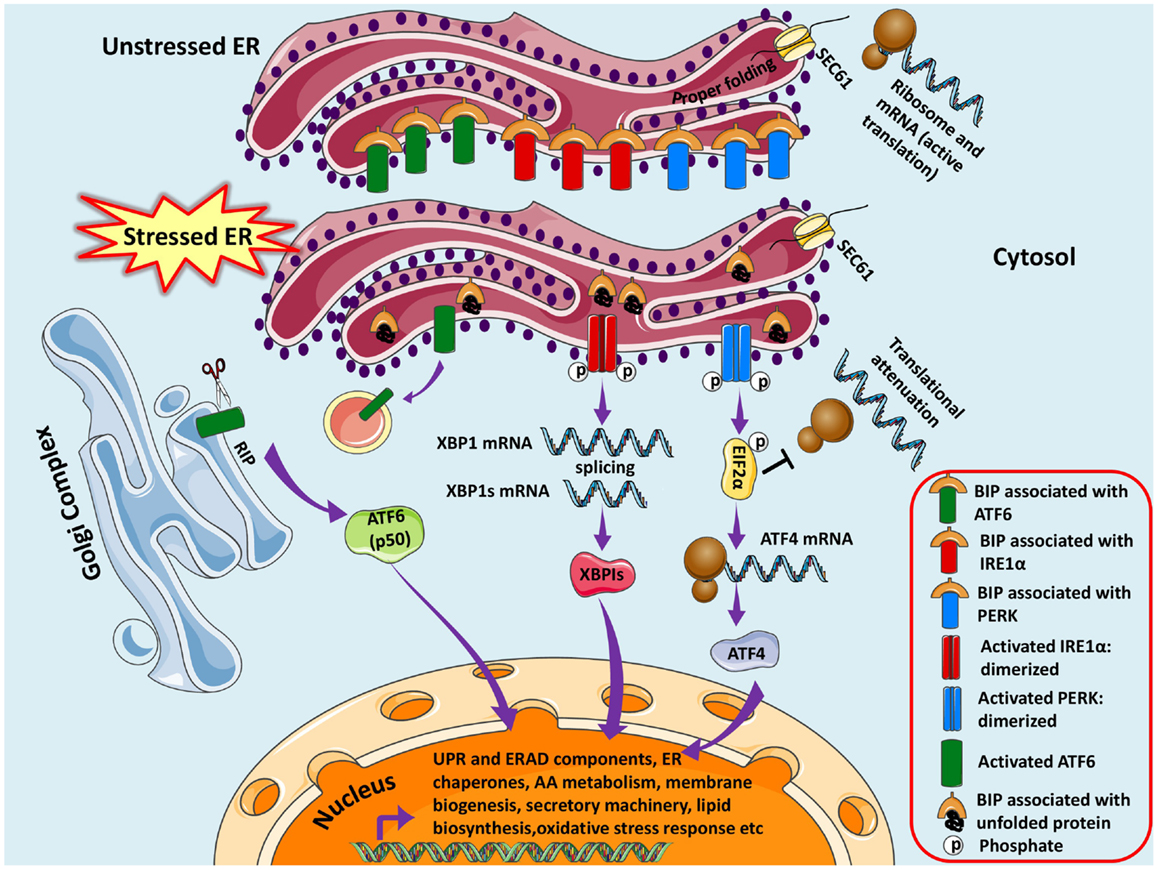 Figure 1. Unfolded protein response pathways are shown. The newly synthesized protein destined for modification in ER enters through SEC61 channel and undergoes folding and maturation. Under basal/unstressed conditions, the BIP (immunoglobulin heavy chain binding protein) binds to ATF6 (activating transcription factor 6), IRE1α (inositol-requiring kinase 1), and PERK [double-stranded RNA-activated protein kinase (PKR)-like endoplasmic reticulum kinase] thereby inhibiting them. During ER stress, BIP dissociates from the three UPR sensors and binds to unfolded/misfolded proteins, thus initiating adaptive signaling events to help ER recover from the stress. Dissociation of BIP from ATF6 unmasks a Golgi-localization signal (GLS, not shown here), which facilitates its translocation to Golgi where it undergoes regulated intramembrane proteolysis (RIP) by resident proteases, Site-1 protease (S1P) and Site-2 protease (S2P). The released ATF6 acts as a transcription factor, which travels to the nucleus and binds to ER-stress response elements (ERSE) and induces transcription of several genes, including BIP, CHOP (CCAAT/enhancer-binding protein homologous protein), and X-box-binding protein 1 (XBP1). Similarly IRE1α is activated and undergoes homodimerization and autophosphorylation, thereby activating the endoribonuclease activity, which splices XBP1 mRNA to spliced XBP1 mRNA, which codes for a transcription factor XBP1s that translocates to nucleus and regulates genes involved in UPR and ER-associated degradation (ERAD). Finally, the release of BIP activates PERK pathway, which initiates a global translational arrest by phosphorylating the translation initiation factor 2α (EIF2α), thus decreasing ER protein load. ATF4 (Activating Transcription Factor 4) mRNA escapes translational suppression exclusively as it possesses internal ribosome entry site (IRES) sequences in the 5′-untranslated regions. ATF4 enters nucleus and regulates expression of UPR target genes. Figure 1. Unfolded protein response pathways are shown. The newly synthesized protein destined for modification in ER enters through SEC61 channel and undergoes folding and maturation. Under basal/unstressed conditions, the BIP (immunoglobulin heavy chain binding protein) binds to ATF6 (activating transcription factor 6), IRE1α (inositol-requiring kinase 1), and PERK [double-stranded RNA-activated protein kinase (PKR)-like endoplasmic reticulum kinase] thereby inhibiting them. During ER stress, BIP dissociates from the three UPR sensors and binds to unfolded/misfolded proteins, thus initiating adaptive signaling events to help ER recover from the stress. Dissociation of BIP from ATF6 unmasks a Golgi-localization signal (GLS, not shown here), which facilitates its translocation to Golgi where it undergoes regulated intramembrane proteolysis (RIP) by resident proteases, Site-1 protease (S1P) and Site-2 protease (S2P). The released ATF6 acts as a transcription factor, which travels to the nucleus and binds to ER-stress response elements (ERSE) and induces transcription of several genes, including BIP, CHOP (CCAAT/enhancer-binding protein homologous protein), and X-box-binding protein 1 (XBP1). Similarly IRE1α is activated and undergoes homodimerization and autophosphorylation, thereby activating the endoribonuclease activity, which splices XBP1 mRNA to spliced XBP1 mRNA, which codes for a transcription factor XBP1s that translocates to nucleus and regulates genes involved in UPR and ER-associated degradation (ERAD). Finally, the release of BIP activates PERK pathway, which initiates a global translational arrest by phosphorylating the translation initiation factor 2α (EIF2α), thus decreasing ER protein load. ATF4 (Activating Transcription Factor 4) mRNA escapes translational suppression exclusively as it possesses internal ribosome entry site (IRES) sequences in the 5′-untranslated regions. ATF4 enters nucleus and regulates expression of UPR target genes.
|
| Paraphrases |
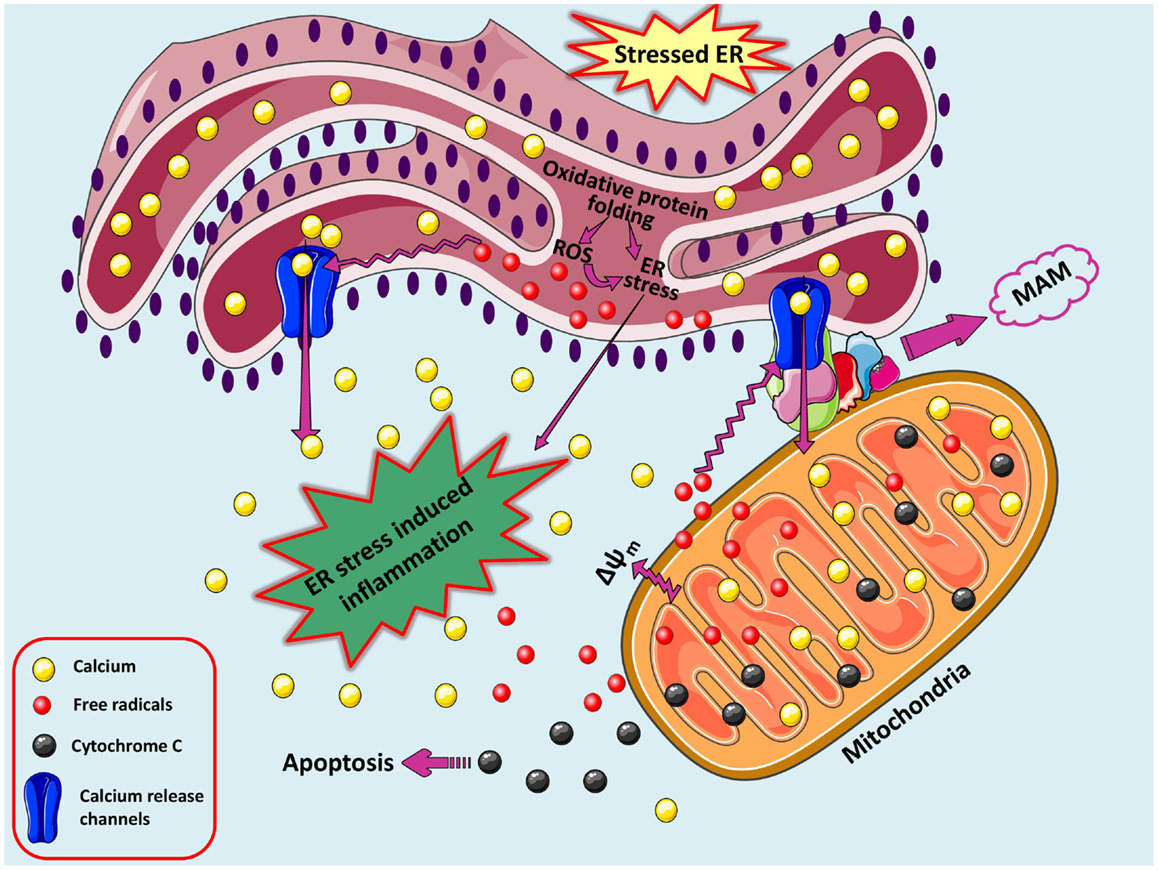 A loop of oxidative stress, ER stress leading to inflammation. During protein overload, ROS are generated in the ER as a part of an oxidative folding process during electron transfer between protein disulfide isomerase (PDI) and endoplasmic reticulum oxidoreductin-1 (ERO-1). ROS can target ER resident proteins, enzymes, and chaperones (not shown) and ER based calcium (Ca2+) channels, leading to the release of calcium from the ER into the cytosol and ER-stress signaling. Increased cytosolic calcium and calcium entry in mitochondria from ER via MAM-associated channels can stimulate mitochondria metabolism to produce more ROS. Increased mitochondrial calcium concentration causes cytochrome c release, altered membrane potential that eventually triggers cellular death programs. The increased protein folding demand, calcium and ROS signaling integrates with UPR pathways and can potentially lead to inflammatory responses. A loop of oxidative stress, ER stress leading to inflammation. During protein overload, ROS are generated in the ER as a part of an oxidative folding process during electron transfer between protein disulfide isomerase (PDI) and endoplasmic reticulum oxidoreductin-1 (ERO-1). ROS can target ER resident proteins, enzymes, and chaperones (not shown) and ER based calcium (Ca2+) channels, leading to the release of calcium from the ER into the cytosol and ER-stress signaling. Increased cytosolic calcium and calcium entry in mitochondria from ER via MAM-associated channels can stimulate mitochondria metabolism to produce more ROS. Increased mitochondrial calcium concentration causes cytochrome c release, altered membrane potential that eventually triggers cellular death programs. The increased protein folding demand, calcium and ROS signaling integrates with UPR pathways and can potentially lead to inflammatory responses.
Added by: Dr. Enrique Feoli
(27/02/2021, 18:26)
|